The BLAST algorithm is a heuristic search method that seeks words of length W (default = 3 in blastp) that score at least T when aligned with the query and scored with a substitution matrix. Words in the database that score T or greater are extended in both directions in an attempt to fina a locally optimal ungapped alignment or HSP (high scoring pair) with a score of at least S or an E value lower than the specified threshold. HSPs that meet these criteria will be reported by BLAST, provided they do not exceed the cutoff value specified for number of descriptions and/or alignments to report. In other word, the BLAST algorithm performs DNA and protein sequence similarity searches by an algorithm that is faster than FASTA but considered to be equally as sensitive. BLAST is very popular due to availability of the program on the World Wide Web through a large server at the National Center for Biotechnology Information (NCBI) and at many other sites. The BLAST algorithm has evolved to provide a set of very powerful search tools for the molecular biologist that are freely available to run on many computer platforms. This article provides a list of steps that describe how the BLAST algorithm searches a sequence database.
PDF about Blast Algorithm https://drive.google.com/open?id=0B3e47MnOSkhWM3hRTWxZVHJwdEk
Key Concepts:
Let's Start
The search space between two sequences can be visualized as a graph with one sequence along the X-axis and the other along the Y-axis (Figure 5-1). Each point in this space represents a pairing of two letters, one from each sequence. Each pair of letters has a score that is determined by a scoring matrix whose values were determined empirically. (See Chapter 4 for more about scoring matrices.) An alignment is a sequence of paired letters that may contain gaps (See Chapter 3 and Chapter 4 for more about gaps). Ungapped alignments appear as diagonal lines in the search space, and the score of an ungapped alignment is simply the sum of the scores of the individual letter pairs. Alignments containing gaps appear as broken diagonals in the search space, and their score is the sum of the letter pairs minus the gap costs, which usually penalize more score points for initiating a gap than extending a gap. How can you tell the difference between two ungapped alignments and a single gapped alignment? In a BLAST report, unaligned regions aren't displayed, and gaps are represented by dashes. However, a simple change in parameters can change one into the other. The diagrams in this chapter show only one gapped alignment, which is indicated in Figure 5-1.
Figure 5-1. Search space and alignment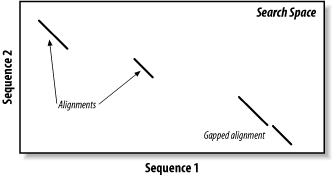
As you saw in Chapter 3, the Smith-Waterman algorithm will find the maximum scoring alignment between two sequences. Some people feel that this ability makes Smith-Waterman the gold standard of alignment algorithms, but this is true only in theory. When comparing real sequences, you may have several good alignments or none. What you really want reported is all of the statistically significant alignments; this is what BLAST does. However, unlike Smith-Waterman, BLAST doesn't explore the entire search space between two sequences. Minimizing the search space is the key to its speed but at the cost of a loss in sensitivity. You will find that the speed/sensitivity trade-off is a key concept when designing BLAST experiments. How exactly does BLAST find similarities without exploring the entire search space? It uses three layers of rules to sequentially refine potential high scoring pairs (HSPs). These heuristic layers, known as seeding, extension, and evaluation, form a stepwise refinement procedure that allows BLAST to sample the entire search space without wasting time on dissimilar regions.
Seeding
BLAST assumes that significant alignments have words in common. A word is simply some defined number of letters. For example, if you define a word as three letters, the sequence MGQLV has words MGQ, GQL, and QLV. When comparing two sequences, BLAST first determines the locations of all the common words, which are called word hits (Figure 5-2). Only those regions with word hits will be used as alignment seeds. This way, BLAST can ignore a lot of the search space.
Figure 5-2. Word hits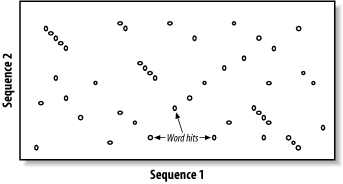
Let's take a moment to define a word hit. A simple interpretation is that a hit is two identical words. Some significant alignments don't contain any identical words, though. Therefore, BLAST employs a more useful definition of a word hit called the neighborhood. The neighborhood of a word contains the word itself and all other words whose score is at least as big as T when compared via the scoring matrix. Therefore, by adjusting T it is possible to control the size of the neighborhood, and therefore the number of word hits in the search space. Table 5-2 shows the neighborhood around the word RGD, and Example 5-1 shows a Perl script for determining the neighborhood for three-letter words.
Table : The neighborhood near RGD
Example : Determining the neighborhood for three-letter words#!/usr/bin/perl -w
use strict;
die "usage: $0 <matrix> <word> <threshold>\n" unless @ARGV == 3;
my ($matrix_file, $WORD, $T) = @ARGV;
my @W = split(//, $WORD);
die "words must be 3 long\n" unless @W == 3;
my @A = split(//, "ARNDCQEGHILKMFPSTWYVBZX*"); # alphabet
my %S; # Scoring matrix
# Read scoring matrix - use those provided by NCBI-BLAST or WU-BLAST
open(MATRIX, $matrix_file) or die;
while (<MATRIX>) {
next unless /^[A-Z\*]/;
my @score = split;
my $letter = shift @score;
for (my $i = 0; $i < @A; $i++) {
$S{$letter}{$A[$i]} = $score[$i];
}
}
# Calculate neighborhood
my %NH;
for (my $i = 0; $i < @A; $i++) {
my $s1 = $S{$W[0]}{$A[$i]};
for (my $j = 0; $j < @A; $j++) {
my $s2 = $S{$W[1]}{$A[$j]};
for (my $k = 0; $k < @A; $k++) {
my $s3 = $S{$W[2]}{$A[$k]};
my $score = $s1 + $s2 + $s3;
my $word = "$A[$i]$A[$j]$A[$k]";
next if $word =~ /[BZX\*]/;
$NH{$word} = $score if $score >= $T;
}
}
}
# Output neighborhood
foreach my $word (sort {$NH{$b} <=> $NH{$a} or $a cmp $b} keys %NH) {
print "$word $NH{$word}\n";
}
The proper value for T depends on both the values in the scoring matrix and the balance between speed and sensitivity. Higher values of Tprogressively remove more word hits (see Figure 5-3) and reduce the search space. This makes BLAST run faster, but increases the chance of missing an alignment.
Figure 3. How T affects seeding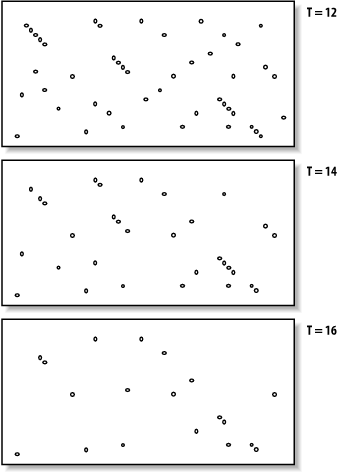
Word size (W) is another variable that controls the number of word hits. It's easy to see why a word size of 1 will produce more hits than a word size of 10. In general, if T is scaled uniformly with W, smaller word sizes increase sensitivity and decrease speed. The interplay between W, T, and the scoring matrix is critical, and choosing them wisely is the most effective way to control the speed and sensitivity of BLAST.
In Figures Figure 2 and Figure 3, you may have noticed that word hits tend to cluster along diagonals in the search space. The two-hit algorithm, as it is known, takes advantage of this property by requiring two word hits on the same diagonal within a given distance (see Figure 5-4). Smaller distances isolate more single word hits and further reduce the search space. The two-hit algorithm is an effective way to remove meaningless, neighborless word hits and improve the speed of BLAST searches.
Figure 4. Isolated and clustered words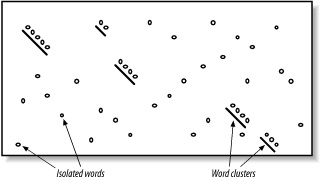 Implementation details
The descriptions thus far have been mostly theoretical. However, some implementation details are worth discussing. In NCBI-BLAST, BLASTN is very different from the other, protein-based algorithms. BLASTN seeds are always identical words; T is never used. To make BLASTN faster, you increase W and to make it more sensitive, you decrease W. The minimum word size is 7. The two-hit algorithm isn't used in BLASTN searches because word hits are generally rare with large, identical words. BLASTP and the other protein-based programs use word sizes of 2 or 3. To make protein searches faster, you set W to 3 and T to a large value like 999, which removes all potential neighborhood words. The two-hit distance is set to 40 amino acids by default, so words don't have to be clustered as closely as they appear in the figures. In principle, setting the two-hit distance to a smaller value also increases speed, but in practice, its effect is insubstantial.
In WU-BLAST, you may set W to any value for any program. If W is set to 5 or more, neighborhood word scores aren't used; they are computed only by explicitly assigning a value for T. High values of W in conjunction with moderate values of T can lead to immense memory requirements, so it is best not to set T when W is 6 or more. To alter the speed/sensitivity of WU-BLAST you can use a variety of combinations of W, and T, and you can also employ the two-hit algorithm.
The statistical model underlying BLAST assumes the letters are independent of one another so that the words MQV and MVQ have the same probability of occurring. However, certain combinations occur in biological sequences much more often than expected. These are usually low-complexity sequences?for example, FFF (see Chapter 2 and Chapter 4). Low-complexity sequences are often of little biological interest and aligning them wastes CPU cycles. Masking these regions is therefore common. Both NCBI-BLAST and WU-BLAST let you replace these regions with N's or X's, which have negative scores when aligned with a nucleotide or amino acid. A more useful technique, termed soft masking, prevents seeding in such regions, but lets alignments extend through them.
Extension
Once the search space is seeded, alignments can be generated from the individual seeds. We've drawn the alignment as arrows extending in both directions from the seeds (Figure 5-5), and you'll see why this is appropriate. In the Smith-Waterman algorithm (Chapter 3), the endpoints of the best alignment are determined only after the entire search space is evaluated. However, because BLAST searches only a subset of the space, it must have a mechanism to know when to stop the extension procedure.
Figure 5-5. Generating alignments by extending seeds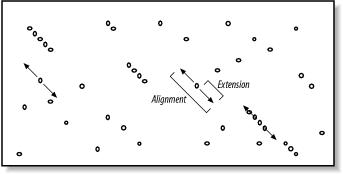
To make this clearer, we'll try aligning two sentences using a scoring scheme in which identical letters score +1 and mismatches score -1. To keep the example simple, ignore spaces and don't allow gaps in the alignment. Although only extension to the right is shown, it also occurs to the left of the seed. Here are the two sentences:
The quick brown fox jumps over the lazy dog.
The quiet brown cat purrs when she sees him.
Assume the seed is the capital T, and you're extending it to the right. You can extend the alignment without incident until you get to the first mismatch:
The quic
The quie
At this point, a decision must be made about whether to continue the alignment or stop. Looking ahead, it's clear that more letters can be aligned, so it would be foolish to stop now. The ends of the sentences, however, aren't at all similar, so we should stop if there are too many mismatches. To do this, we create a variable, X, that represents the recent alignment history. Specifically, X represents how much the score is allowed to drop off since the last maximum. Let's set X to 5 and see what happens. We'll keep track of the sum score and the drop off score as we go. Figure 5-6 shows the graphical interpretation.
The quick brown fox jump
The quiet brown cat purr
123 45654 56789 876 5654 <- score
000 00012 10000 123 4345 <- drop off score
Figure 5-6. Attenuating extension with X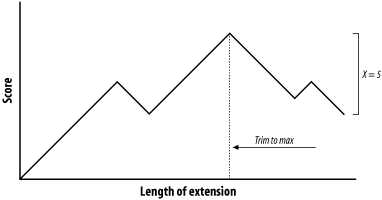
The maximum score for this alignment is 9, and the extension is terminated when the score drops to 4. After terminating, the alignment is trimmed back to the maximum score. If you set X to 1 or 2, the best alignment has a score of 6. If you set X to 3, you can retrieve the longer alignment and save a little computation. A very large value of X doesn't increase the score and requires more computation. So what is the proper value for X? It's generally a good idea to use a large value, which reduces the risk of premature termination and is a better way to increase speed than with the seeding parameters. However, W, I, and 2-hit are better for controlling speed than X.
The gapless extension algorithm just demonstrated is similar to what was used in the original version of BLAST. The algorithms in the current versions of BLAST allow gaps and are related to the dynamic programming techniques described in Chapter 3. Therefore, X not only depends on substitution scores, but also gap initiation and extension costs.
Implementation detailsExtension in BLASTN is a little different from BLASTP and the other protein-based programs. The reason for this has to do with how nucleotide sequences are stored in BLAST databases. Because there are only four common nucleotide symbols, nucleotide databases can be stored in a highly compressed state with only two bits per nucleotide. What happens if the sequence contains an N or other ambiguous nucleotide? A random canonical nucleotide is substituted. For example, an N can be randomly changed to A, C, G, or T and a Y changed to a C or T. It is possible, especially with long stretches of ambiguous nucleotides, that the two-bit approximation terminates extension prematurely.
NCBI-BLAST and WU-BLAST take very different approaches to the gapped extension procedure. NCBI-BLAST has three values for X (parameters -X-y -Z) and WU-BLAST has two (parameters -X and -gapX). Some differences, such as the presence of a floating bandwidth (NCBI) rather than a fixed bandwidth, are interesting from an academic viewpoint but less so from a user's perspective. What is important is that altering the extension parameters from their defaults is generally not an effective way to increase the speed or sensitivity of a BLAST search. You might consider adjusting the parameters in two situations:
Since gapped extension also depends on the gap initiation and extension costs, you should note that these parameters are interpreted differently in NCBI-BLAST and WU-BLAST. In NCBI-BLAST, the total cost for a gap is the gap opening cost (-G) plus the gap extension cost (-E) times the length of the gap. In WU-BLAST, the total cost of a gap is the cost of the first gap character (-Q) plus all remaining gap characters (-R). The NCBI parameters -G 1 -E 1 are identical to -Q 2 -R 1 in WU-BLAST.
Evaluation
Once seeds are extended in both directions to create alignments, the alignments are evaluated to determine if they are statistically significant (Chapter 4). Those that are significant are termed HSPs. At the simplest level, evaluating alignments is easy; just use a score threshold, S, to sort alignments into low and high scoring. Because S and E are directly related through the Karlin-Altschul equation, a score threshold is synonymous with a statistical threshold. In practice, evaluating alignments isn't as simple, which is due to complications that result from multiple HSPs.
Consider the alignment between a eukaryotic protein and its genomic source. Because most coding regions are broken up by introns, an alignment between the protein and the DNA is expected to produce several HSPs, one for each exon. In assessing the statistical significance of the protein-DNA match, should each exon alignment be forced to stand on its own against the statistical threshold, or does it make more sense to combine the scores of the various exons? The latter is generally more appropriate, especially if some exons are short and may be thrown out if not aided in some way. However, determining the significance of multiple HSPs isn't as simple as summing all the alignment scores because many alignments are expected to be extensions from fortuitous word hits and not all groups of HSPs make sense.
An alignment threshold is an effective way to remove many random, low-scoring alignments (Figure 5-7). However, if the threshold is set too high, (Figure 5-7c), it may also remove real alignments. This alignment threshold is based on score and therefore doesn't consider the size of the database. There are, of course, E-value and P-value interpretations, if you consider the size of individual sequences or a constant theoretical search space.
Figure 5-7. Increasing alignment thresholds remove low scoring alignments
Qualitatively, the relationship between HSPs should resemble the relationship between ungapped alignments. That is, the lines in the graph should start from the upper left and continue to the lower right, the lines shouldn't overlap, and there should be a penalty for unaligned sequence. Groups of HSPs that behave this way are considered consistent. Figure 5-8 shows consistent and inconsistent HSPs. From a biological perspective, you expect the 5´ end of a coding sequence to match the N-terminus of a protein and the 3´ end to match the C-terminus?not vice versa.
Figure 5-8. Consistent and inconsistent alignment groups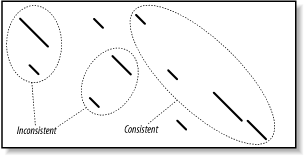
The algorithm for defining groups of consistent HSPs compares the coordinates of all HSPs to determine if there are overlaps (a little overlap is actually allowed to account for extensions that may have strayed too far). This computation is quadratic in the number of HSPs and therefore can be costly if there are many HSPs (e.g., when the sequences are long, and the alignment threshold is low).
Once HSPs are organized into consistent groups, they can be evaluated with a final threshold based on the entire search space and corresponding to the value of E set for the search. You can read more about this topic in Chapter 4. BLAST reports any alignment or group of alignments that meets the E requirement.
Implementation details
This chapter initially described BLAST as having three phases: seeding, extension, and evaluation. In reality, BLAST isn't so straightforward. There are two rounds of extension and evaluation: ungapped and gapped. Gapped extension and evaluation are triggered only if ungapped alignments surpass the ungapped thresholds. In other words, to find a gapped alignment, you must first find a reasonable ungapped alignment.
In NCBI-BLAST, the command line parameter -e sets the final threshold. The value for the alignment threshold is set by the software and isn't a user-definable parameter. You can find the value for E in the footer. For example, if E is set with -e 1e-10, E is reported as follows:
Number of sequences better than 1.0e-10: 4
The value for the alignment threshold and its gapped equivalent are displayed respectively as S1 and S2 with the raw score listed first. Note that the ungapped threshold is quite a bit lower than the gapped threshold.
S1: 41 (21.7 bits)
S2: 158 (65.5 bits)
In WU-BLAST, the E or S parameters specify the final threshold (if both are specified, the most stringent one is used). The command-line parameters S2 and its gapped counterpart gapS2 specify the alignment threshold. WU-BLAST includes E-value versions of the alignment threshold based on a constant search space. They may be set via E2 and gapE2. The values for these parameters are shown in the footer. In this example, the alignment threshold (S2) has an ungapped threshold of 33 and a gapped threshold of 36 (one line below).
Query
Frame MatID Length Eff.Length E S W T X E2 S2
+0 0 235 235 10. 65 3 12 22 0.20 33
32 0.21 36
While gapped alignments are useful from a biological perspective, they pose a small problem to Karlin-Altschul statistics because there is no known way to calculate lambda with arbitrary gap penalties. However, lambda can be estimated by observing the properties of random alignments in a given scoring scheme. Both NCBI-BLAST and WU-BLAST have internal tables that contain Karlin-Altschul parameters for common matrices and gap penalties. If you try to use an unsupported scoring scheme in NCBI-BLAST, the program will terminate and list the possible gap penalties. Unsupported scoring schemes in WU-BLAST revert to ungapped parameters, but a warning is issued that informs you that you can provide your own values for lambda, k, and H on the command line.
NCBI's version of BLASTN doesn't contain gapped values for lambda; lambda is always calculated directly from the match/mismatch scores. Because of this, equivalent alignments may have much higher bit scores (and lower E-values) in NCBI-BLAST than WU-BLAST, even if their match/mismatch scores are identical.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Saturday, March 18, 2017
The Blast Algorithm

Why Microsoft is calling Windows 10 is 'the last version of Windows?

IT'S ALL ABOUT WINDOWS AS A SERVICE
Microsoft has been discussing the idea of Windows as a service, but the company hasn't really explained exactly how that will play out with future versions of Windows. That might be because there won't really be any future major versions of Windows in the foreseeable future. Microsoft has altered the way it engineers and delivers Windows, and the initial result is Windows 10. Instead of big releases, there will be regular improvements and updates. Part of this is achieved by splitting up operating system components like the Start Menu and built-in apps to be separate parts that can be updated independently to the entire Windows core operating system. It's a big undertaking, but it's something Microsoft has been actively working on for Windows 10 to ensure it spans across multiple device types.
While we'll witness the results in the coming months, Microsoft is already in launch mode for a number of its apps and services that power Windows 10. The software company is testing preview builds of Window 10 with willing participants, and apps like Xbox and Mail have been engineered for regularly monthly updates. Even Office for Windows 10 will also get regular updates, much like a mobile version, instead of the big bang release every few years.
WINDOWS ISN'T DEAD, BUT THE IDEA OF VERSION NUMBERS COULD BE
When I reached out to Microsoft about Nixon's comments, the company didn't dismiss them at all. "Recent comments at Ignite about Windows 10 are reflective of the way Windows will be delivered as a service bringing new innovations and updates in an ongoing manner, with continuous value for our consumer and business customers," says a Microsoft spokesperson in a statement to The Verge. "We aren’t speaking to future branding at this time, but customers can be confident Windows 10 will remain up-to-date and power a variety of devices from PCs to phones to Surface Hub to HoloLens and Xbox. We look forward to a long future of Windows innovations."
With Windows 10, it's time to start thinking of Windows as something that won't see a big launch or major upgrade every few years anymore. Much like how Google's Chrome browser gets updated regularly with version numbers nobody really pays attention to, Microsoft's approach will likely result in a similar outcome. This is really the idea of Windows as a service, and the notion that Windows 10 could be the last major version of Windows. Microsoft could opt for Windows 11 or Windows 12 in future, but if people upgrade to Windows 10 and the regular updates do the trick then everyone will just settle for just "Windows" without even worrying about the version number.
What is Life?

Introduction
In the intro to biology video, we defined biology as the branch of science concerned with the study of living things, or organisms. That definition is pretty straightforward. However, it opens the door to more difficult—and more interesting—questions: What is life? What does it mean to be alive?
You are alive, and so am I. The dog I can hear barking is alive, and so is the tree outside my window. However, snow falling from the clouds is not alive. The computer you’re using to read this article is not alive, and neither is a chair or table. The parts of a chair that are made of wood were once alive, but they aren’t any longer. If you were to burn the wood in a fire, the fire would not be alive either.
What is it that defines life? How can we tell that one thing is alive and another is not? Most people have an intuitive understanding of what it means for something to be alive. However, it’s surprisingly hard to come up with a precise definition of life. Because of this, many definitions of life are operational definitions—they allow us to separate living things from nonliving ones, but they don’t actually pin down what life is. To make this separation, we must come up with a list of properties that are, as a group, uniquely characteristic of living organisms.
Properties of life
Biologists have identified various traits common to all the living organisms we know of. Although nonliving things may show some of these characteristic traits, only living things show all of them.
1. Organization
Living things are highly organized, and all living organisms are made up of one or more cells, which are considered the fundamental units of life. Individual cells perform complex biochemical processes needed to maintain their structure and function, and each cell is highly organized.
Unicellular organisms consist of only a single cell, while multicellular organisms—such as humans—are made up of many cells. The cells in multicellular organisms may be specialized to do different jobs and are organized into tissues, such as connective tissue, epithelial tissue, muscle, and nervous tissue. Tissues make up organs, such as the heart or lungs, which carry out specific functions needed by the organism as a whole.

Image credit: left, modified from "Prokaryote cell by Ali Zifan (CC BY-SA 4.0), modified image is licensed under a CC BY-SA 4.0 license; center, modified from "Four types of tissue" by the National Institutes of Health (public domain); rIght, modified from "PseudostratifiedCiliatedColumnar" by Blausen staff (CC BY 3.0)
2. Metabolism
Life depends on an enormous number of interlocking chemical reactions. These reactions make it possible for organisms to do work—such as moving around or catching prey—as well as growing, reproducing, and maintaining the structure of their bodies. Living things must use energy and consume nutrients to carry out the chemical reactions that sustain life. The sum total of the biochemical reactions occurring in an organism is called its metabolism.
Metabolism can be subdivided into anabolism and catabolism. In anabolism, organisms make complex molecules from simpler ones, while in catabolism, they do the reverse. Anabolic processes typically consume energy, whereas catabolic processes can make stored energy available.
3. Homeostasis
Living organisms regulate their internal environment to maintain the relatively narrow range of conditions needed for cell function. For instance, your body temperature needs to be kept relatively close to 98.6F (37C). This maintenance of a stable internal environment, even in the face of a changing external environment, is known as homeostasis.

4. Growth
Living organisms undergo regulated growth. Individual cells become larger in size, and multicellular organisms accumulate many cells through cell division. You yourself started out as a single cell and now have tens of trillions of cells in your body! Growth depends on anabolic pathways that build large, complex molecules such as proteins and DNA, the genetic material.
5. Reproduction
Living organisms can reproduce themselves to create new organisms. Reproduction can be either asexual, involving a single parent organism, or sexual, requiring two parents. Single-celled organisms, like the dividing bacterium shown in the left panel of the image at right, can reproduce themselves simply by splitting in two!

In sexual reproduction, two parent organisms produce sperm and egg cells containing half of their genetic information, and these cells fuse to form a new individual with a full genetic set. This process, called fertilization, is illustrated in the image at far right.
6. Response
Living organisms show “irritability,” meaning that they respond to stimuli or changes in their environment. For instance, people pull their hand away—fast!—from a flame; many plants turn toward the sun; and unicellular organisms may migrate toward a source of nutrients or away from a noxious chemical.

7. Evolution
Populations of living organisms can undergo evolution, meaning that the genetic makeup of a population may change over time. In some cases, evolution involves natural selection, in which a heritable trait, such as darker fur color or narrower beak shape, lets organisms survive and reproduce better in a particular environment. Over generations, a heritable trait that provides a fitness advantage may become more and more common in a population, making the population better suited to its environment. This process is called adaptation.
Is this the definitive list?
Living organisms have many different properties related to being alive, and it can be hard to decide on the exact set that best defines life. Thus, different thinkers have developed different lists of the properties of life. For instance, some lists might include movement as a defining characteristic, while others might specify that living things carry their genetic information in the form of DNA. Still others might emphasize that life is carbon-based.
Nonetheless, the list above provides a reasonable set of properties to help us distinguish between things that are alive and those that are not.
Separating living and non-living things
How well do the properties above allow us to determine whether or not something is alive? Let’s revisit the living and nonliving things we saw in the introduction as a test.
The living things we saw in the introduction—humans, dogs, and trees—easily fulfill all seven criteria of life. We, along with our canine friends and the plants in our yards, are made of cells, metabolize, maintain homeostasis, grow, and respond. Humans, dogs, and trees are also capable of reproducing, and their populations undergo biological evolution.
Nonliving things may show some, but not all, properties of life. For instance, crystals of snow are organized—though they don't have cells—and can grow but don’t meet the other criteria of life. Similarly, a fire can grow, reproduce by creating new fires, and respond to stimuli and can arguably even be said to “metabolize.” However, fire is not organized, does not maintain homeostasis, and lacks the genetic information required for evolution.
Living things may keep some properties of life when they become nonliving, but lose others. For instance, if you looked at the wood in a chair under a microscope, you might see traces of the cells that used to make up the living tree. However, the wood is no longer alive, and, having been made into a chair, can no longer grow, metabolize, maintain homeostasis, respond, or reproduce.
What counts as life is still being defined.
The question of what it means to be alive remains unresolved. For instance, viruses—tiny protein and nucleic acid structures that can only reproduce inside host cells—have many of the properties of life. However, they do not have a cellular structure, nor can they reproduce without a host. Similarly, it’s not clear that they maintain homeostasis, and they don’t carry out their own metabolism.

Image credit: Image modified from "Enveloped icosahedral virus," by Anderson Brito (CC BY-SA 3.0; the modified image is licensed under a CC BY-SA 3.0 license
For these reasons, viruses are not generally considered to be alive. However, not everyone agrees with this conclusion, and whether they count as life remains a topic of debate. Some even simpler molecules, such as self-replicating proteins—like the “prions” that cause mad cow disease—and self-replicating RNA enzymes, also have some, but not all, of the properties of life.
Moreover, all of the properties of life we have discussed are characteristic of life on earth. If extraterrestrial life exists, it may or may not share the same characteristics. Indeed, NASA’s working definition that “life is a self-sustaining system capable of Darwinian evolution” opens the door to many more possibilities than the criteria defined above. However, this definition also makes it hard to quickly decide whether something is alive!
As more types of biological entities are discovered, on Earth or beyond, they may demand that we re-think what it means for something to be alive. Future discoveries may call for revisions and extensions of the definition of life.
What do you think?
How would you define life? Would you add something to the list of properties above, subtract something, or use an entirely different definition? Can you think of exceptions or special cases that aren’t covered by the list? Share your ideas in the comments section below!





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}